Simulating Ferri/Ferrocyanide Reaction in Julia
Update: This post uses a deprecated library. See Ferri/Ferrocyanide Reaction with diffusion in Julia – Part 2 for a version, which uses non-deprecated stuff. A least for now…
I have lately been playing around with differential equations (PDEs) in the programming language Julia. Prior to this exercise, PDEs and ODEs (Ordinary Differential Equations) where things I tried to avoid at University. But nowadays they have caught my interest.
The Ferri/Ferrocyanide reaction is a well studied reaction in the area of Electrochemistry (i.e. all chemistry which involves the exchange of electrons). The reaction is specifically used in combination with a method called Cyclic Voltammetry.
This exercise is my attempt to increase my knowledge on both subjects. So…
Disclaimer: I am no electrochemist nor have any experience with solving differential equations. There might be some mistakes in the text below…
The simulation works by solving the following diffusion equation:
Where ci(x,t) is the concentration of molecule i at position x at time t. The boundary condition is determined by a redox reaction (in this case the ferri/ferrocyanide reaction). It is assumes that we have a cell where position x=0 is the active electrode.
If we have a redox reaction:
Then the reaction rate r(t) at time is defined as:
Here kf is the forward constant rate and kb is the backward constant rate. The concentration of the to be oxidised molecule is co and the concentration of the to be reduced molecule is cr.
The rates kf and kb can be computed using:
Here E(t) is the applied potential at time t. E₀ is equilibrium potential for the reaction couple. k0 is the standard heterogeneous rate constant for the reaction couple. n is the amount of electrons involved in the reaction. In this case it is 1, so we can omit n for the equation. The temperature needs to be 298.15K since this is the temperature for which E0 and k0 are defined. Finally we have the Faraday constant F and the gas constant R.
To speed up computation and increase readability, I define:
Such that:
Finally the current i(t) at time t can be computed using:
Where A is the surface of the electrode.
Ferri/ferrocyanide reaction formula
The concrete reaction we’re looking at is:
Where the lefthand side is called ferricyanide and the righthand side is called ferrocyanide.
First bit of the program: The external packages
First of all, some external packages need to be included.
using DifferentialEquations
using DiffEqOperators
using RecursiveArrayTools
using Gadfly
using Catalyst
using SymbolicsDifferentialEquations is the main provider of functionality to solve differential equations. DiffEqOperators provides functionality to transform PDEs into ODEs. At the moment I’m writing this article, the package is already deprecated. So I might post an update some day.
RecursiveArrayTools provides ArrayPartition. I’ll get back on that one later. Symbolics allows symbolic modelling. Catalyst allows symbolic modelling specifically for chemical reactions. Finally, there is Gadfly, which is used for plotting.
Constants
There are a lot of constants is the program. I prefer to define them all at a single place. It is common to use unicode in Julia. This allows the source code to stay closer to the actual equations. Annoyingly enough, not every character is easily supported. Subscripts ‘f’ and ‘b’ are missing for example. I’ve replaced them by an ‘a’ and a ‘β’ subscript (a ‘b’ subscript is also not supported).
const width = 0.01 # The width of the cell
const Δx = 0.0002 # The distance between points in the cell
const Nₓ = floor(Int, width/Δx) # The amount of discrete points in the cell
const A = 1e-6
const F = 96485.3329 # Faraday constant
const R = 8.31446261815324 # Universal gas constant
const E₀ = 0.37 # standard electrode potential
const k₀ = 2.5e-4 # reaction rate constant
const T = 298.15 # Temperture
const α = 0.5
const λₐ = (-α*F)/(T*R)
const λᵦ = ((1-α)*F)/(T*R)
const Dₒ = 0.6e-9 # Diffusion coefficient for ferrocyanide
const Dᵣ = 0.7e-9 # Diffusion coefficient for ferricyanide
const iₒ = 1
const iᵣ = 2Split ODE problem
The overall approach is that a so-call Split ODE problem is defined. A split ODE problem is nothing more than that the ODE problem is formulated using 2 functions:
In this case the equation has one linear element and is of the form:
Where the linear part represents the diffusion part and f is the reaction part.
Diffusion
Both the diffusion of ferricyanide as well as ferrocyanide needs to be defined. Before showing the actual code, let’s first take a few steps back.
For normal diffusion processes Fick’s law seems to be the foundation. Ficks first equation is defined as:
where
- The diffusion flux J is the amount of molecules flowing through and area within a certain time period.
- The diffusion coefficient D is assumed to be constant and something we need to get from some table.
- The concentration of a molecule is 𝜑.
- The position along the gradient is x.
For 2D or more we need the notation, where J is now a flux vector.:
Fick’s law can be turned into the Diffusion Equation by replacing 𝜑 with the function c(x,t), which provides the concentration of a molecule at position x and time t:
One approach to solve the PDE is by discretising the 1D space into small cells with fixed width Δx. The second derivative relative to the distance is defined as:
at any non-boundary position x at any given time t. This can also be written in matrix form as:
Where u is the vector of all concentrations and the matrix A is a diagonal matrix as shown below.
Multiply the resulting derivative vector du with D to get the change in concentration within 1 second. Add du * D to vector u to get the new concentrations. All this we can tie together to create a step function:
using LinearAlgebra
n = 50
Δx = 0.001
D = 1e-9
u₀ = zeros(50)
u₀[5:10] .= 1
A = SymTridiagonal(fill(-2.0, length(ϕ₀)), ones(length(ϕ₀)-1))
step(u, D, Δx) = ϕ .+ (A * u) .* (D/(Δx^2))
u₁ = step(u₀, D, Δx)
u₂ = step(u₁, D, Δx)If you want to take different time steps than 1s, you need to multiply the derivative vector with some Δt as well.
In the actual program the matrix is created using some tools from DiffEqOperators, which creates one ready to use matrix with all the whistles and bells.
Δ = CenteredDifference(2, 2, Δx, 50)
bc = Neumann0BC(Δx)
Δₒ = Dₒ * Δ * bc
Δᵣ = Dᵣ * Δ * bc
In the code above, the boundary conditions are also defined. Two functions are provided by DiffEqOperators :
- Dirichlet: Set the boundary concentration certain value. For example when the boundary of a solution is the container, then the concentration is always 0.
- Neumann: Set the boundary flux to a certain value. For example the ions created by an oxidation reaction with an electrode.
The final thing we need to do, is to combine the concentrations for Ferro- and ferrocyanide in one single vector. This is done by using an ArrayPartition as type for u.
When we write this all in a form suitable for an ODE solver, we get:
function diffusion!(du, u::ArrayPartition, _, t)
uₒ = u.x[iₒ]
uᵣ = u.x[iᵣ]
du.x[iₒ] .= Δₒ * uₒ
du.x[iᵣ] .= Δᵣ * uᵣ
endReaction
The reaction rate equation described above is implemented as follows:
function redox_rate(cₒ, cᵣ, E)
η = E - E₀
kₐ = k₀ * exp(λₐ * η)
kᵦ = k₀ * exp(λᵦ * η)
kₐ * cₒ - kᵦ * cᵣ
endThe actual reaction function for the solver:
function reaction!(du, u, p, t)
du .= 0.0
E = p[1]
cₒ = u.x[iₒ][1]
cᵣ = u.x[iᵣ][1]
r = redox_rate(cₒ, cᵣ, E(t))
du.x[iₒ][1] = -r
du.x[iᵣ][1] = r
endE is a function which provides the potential at time t. For each created ferrocyanide molecule one ferricyanide molecule is destroyed. This means that the reaction rate for ferricyanide is the opposite of the reaction rate of ferrocyanide.
Solving the split ODE problem
function run(u₀::ArrayPartition, t_max, E; savelog=false)
p = [E]
prob = SplitODEProblem(diffusion!, reaction!, u₀, (0.0, t_max), p)
alg = BS3()
s = savelog ? [10^i for i ∈ 0:0.2:log10(t_max)] : (1:(t_max/200):t_max)
solve(prob, alg, saveat=s, save_start=true, abstol=1e-10, reltol=1e-10)
endAbove the split ODE problem is defined together with the vector u0, which contains the initial concentrations of Ferro- and ferrocyanide in each cell. The function E is provided as parameter. The problem runs from 0 to t_max seconds. The array s defines at which times data needs to be saved, such that we have something nice to plot.
Selecting a good solver is still a little black magic for me. The documentation provides some good starting points. From there I just experimented with several solvers to see which ones performed best. I ended up with BS3, although in many cases ROCK4 is much faster. But sometimes the plots look less smooth. Not sure why that is…
About speed…running the solver with BS3 takes about 5.6s on my computer, and with ROCK4 it takes 1.5s. But only if I run it directly in the REPL. I prefer to develop using Pluto, which is an awesome notebook system…except with the latest versions in combination with solving differential equations. Running the solver from a Pluto notebook takes about half an hour. It used to be similar to running it directly in the REPL, but some reason it slowed down a lot. No clue yet why…
Using Catalyst
The ferri/ferrocynaide reaction is fairly simple. If we ever want to solve a more complicated reaction, then Catalyst seems to provide much better tools define the reaction function. Catalyst allows a developer to symbolically define and ODE using actual chemical equations and reaction rate constants.
function run_with_catalyst(u₀::ArrayPartition, t_max, E, saveat=1)
# Function to compute the reaction rate constants
kb(k₀, E₀, E, α) = k₀ * exp(((-α * F)/(T*R)) * (E - E₀))
kf(k₀, E₀, E, α) = k₀ * exp((((1-α) * F)/(T*R)) * (E - E₀))
# Define the reaction system at the electrode
sys_electrode = @reaction_network begin
(kb(k1, E1, E(t), α1), kf(k1, E1, E(t), α1)), FeCN₆¯³ ⟷ FeCN₆¯⁴
end k1 E1 α1
# Convert the system such that we can combine it with the diffusion
f_electrode = ODEFunction(convert(ODESystem, sys_electrode))
# The reaction at the electrode
function _reaction!(du, u, p, t)
du .= 0.0
u2 = zeros(2)
u2[1] = u.x[1][1]
u2[2] = u.x[2][1]
du2 = zeros(2)
f_electrode.f(du2, u2, p, t)
du.x[1][1] = du2[1]
du.x[2][1] = du2[2]
end
p = [k₀, E₀, α]
prob = SplitODEProblem(diffusion!, _reaction!, u₀, (0.0, t_max), p)
alg = BS3()
solve(prob
, alg
, saveat=saveat
, save_start=true
, abstol=1e-10
, reltol=1e-10
, maxiters=1e7
)
endRunning CV
Cyclic Voltammetry (CV) is a method, where a voltage is applied over an electrochemical cell. The voltage is increased with a certain rate until a maximum is reached. After the that the voltage is decreased with the same rate until a lower limit is reach. Then it is increased again, etc…
The resulting potential vs time plot looks something like this:
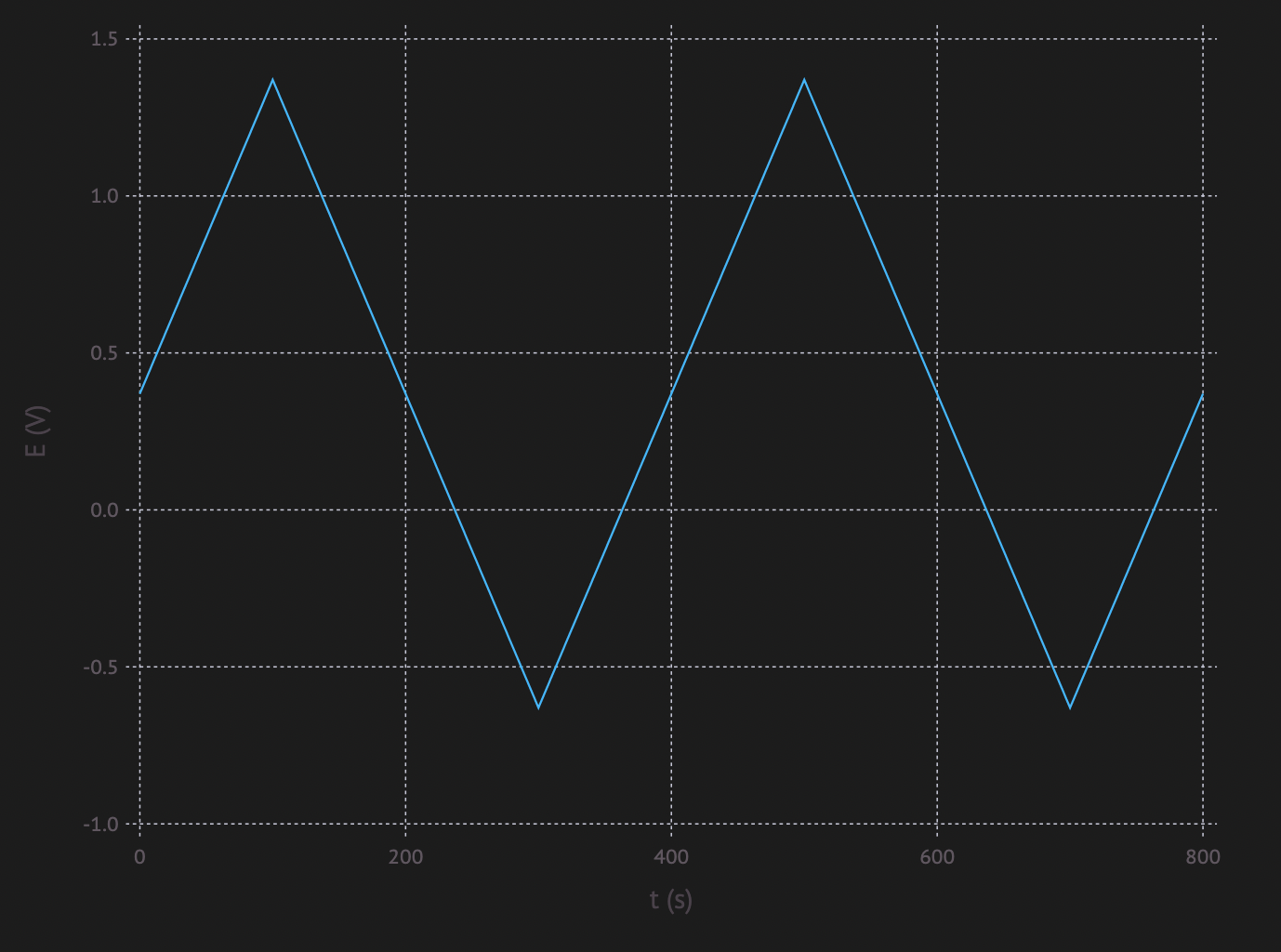
This causes the reaction to take place and reverse all the time. One of the things one can analyse with this, is if a reaction is fully reversible.
The function below generates the CV potentials for each time t.
function cv(t)
s = 0.01
t₁ = inv(s)
if t < t₁
s * t + E₀
else
i = floor(Int, (t - t₁)/(2t₁))
if iseven(i)
-s * (t - (2i+1)*t₁) + E₀ + 1.0
else
s * (t - (2i+1)*t₁) + E₀ - 1.0
end
end
endIn order to use the cv function with Catalyst, it needs to be registered:
@register cv(t)From the concentrations at each point in time, the reaction rate can be computed. Using the reaction rate, we can compute the current.
function current(sol, E)
Cₒ = [u.x[iₒ][1] for u ∈ sol.u]
Cᵣ = [u.x[iᵣ][1] for u ∈ sol.u]
R = redox_rate.(Cₒ, Cᵣ, E.(sol.t))
b = F * A
b .* R
endA function to create a plot:
function plot_I_vs_E(I, E)
plot(x=E, y=I
, Guide.xlabel("E (V)")
, Guide.ylabel("I (A)")
, Guide.title("I vs E")
, Geom.line(preserve_order=true)
, Theme(default_color="purple")
)
endRun the solver, convert the outcome to current and plot it:
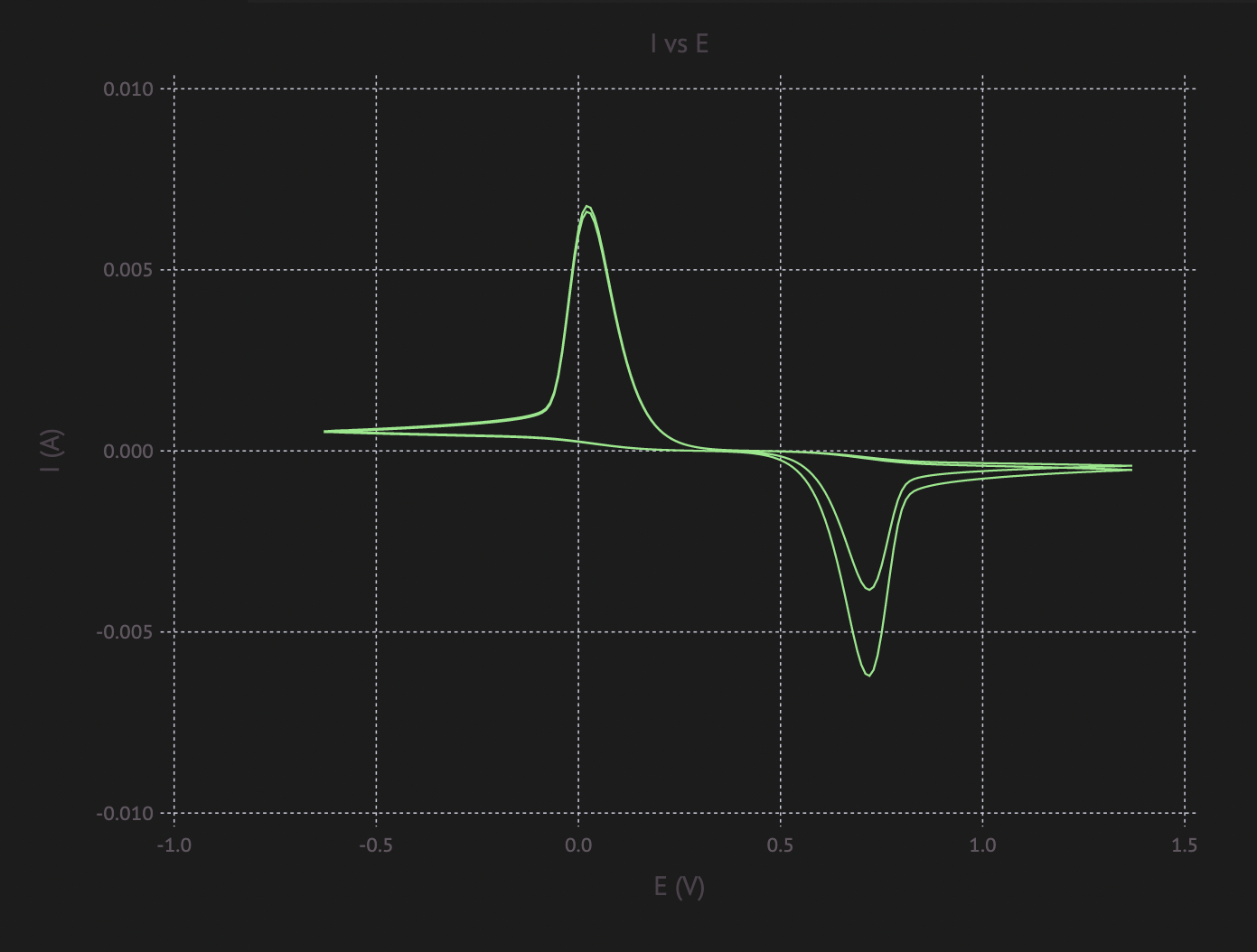
sol = run_with_catalyst(ArrayPartition(fill(0.5, Nₓ), fill(0.5, Nₓ)), 800, cv)
plot_I_vs_E(current(sol, cv), cv.(sol.t))And the final picture looks somewhat like what you might expect from running CV (example from research paper):